Our research focuses on advancing cancer treatments through the development of innovative methods and the analysis of complex data sets.
#spatialbiology #computationalbiology #tumorecosystem
We conduct research in the following major areas:
Melanoma, a type of skin cancer, is a highly aggressive and potentially fatal disease. While there has been significant progress in the treatment of melanoma, it remains a major public health concern. In recent years, the role of the tumor microenvironment, which is the cellular environment that surrounds the tumor, in driving the development and progression of melanoma has come to focus. Specifically, the restructuring of the tumor microenvironment has emerged as a key factor in promoting the growth and spread of melanoma. By gaining a fundamental understanding of this process, we hope to identify new targets for the development of more effective melanoma treatments.
Our Approach:
We study the role of tumor microenvironment restructuring in melanoma development by integrating high-dimensional datasets such as multiplexed imaging, high-resolution 3D imaging, single-cell RNA sequencing data, and genomics data. These tools allow us to gain a more comprehensive understanding of the complex cellular interactions that drive melanoma progression and identify potential targets for more effective treatment.
Our Discoveries:
Early spatial changes in microenvironment predict Tumor Progression: Our investigation focused on how the tumor microenvironment (TME) is remodeled during the early stages of melanoma development. We found that although the overall immune cell composition remained relatively constant between normal tissue and early tumor-precursors, the spatial distribution of these immune cells was altered. These results suggest that microenvironmental changes occur very early on in the development of melanoma, and may be critical in driving the progression of the disease.
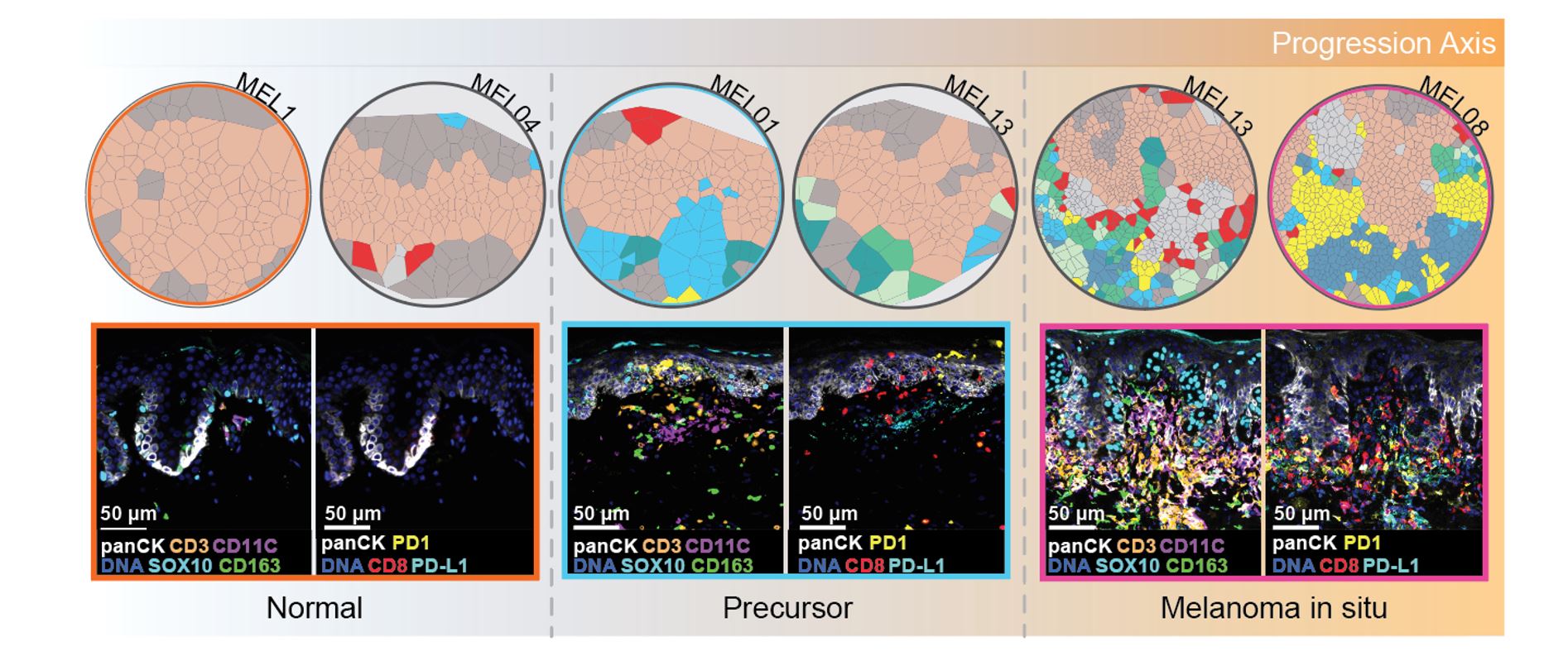
An atlas of changes observed in the tumor microenvironment (TME) during the progression of melanoma: Our data indicate that as the disease advances, immune cells exhibit altered spatial distribution within the TME, which is characterized by both immune activation and suppression. Specifically, during the transition from normal tissue to the precursor stage of melanoma, we observed the migration of cytotoxic T cells towards melanocytes, accompanied by partial migration of regulatory T cells and PDL1+ dendritic cells. These findings suggest the early activation of immune suppression in the TME. In the melanoma in situ stage, we noted that the cytotoxic T cells were located further away from the melanocytes, while the formation of a myeloid niche in close proximity to the melanocytes was prominent. This observation suggests the presence of a highly immune-suppressed microenvironment. Finally, during the transition to invasive melanoma, we observed pronounced and consolidated effects, with myeloid cells located close to tumor cells expressing PDL1 and most CD8 T cells expressing markers of exhaustion. Taken together, our findings highlight the dynamic changes that occur in the TME during melanoma progression, and provide insight into the development of immunosuppression in the microenvironment.
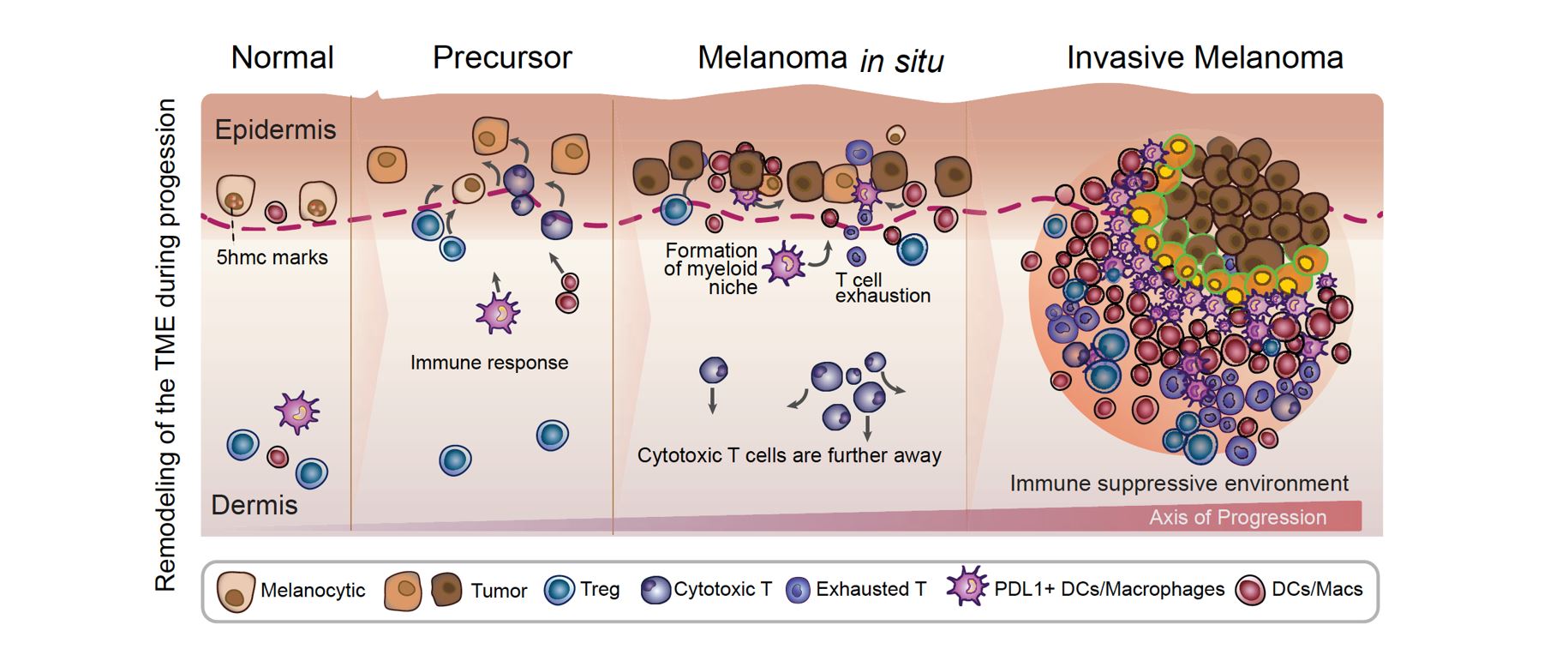
The tumor microenvironment (TME) is a critical factor in the development, progression, and treatment of melanoma. We aim to gain a deeper understanding of the communication between cells within the TME, unlocking new insights into the mechanisms that drive tumor growth, invasion, and metastasis.
Our Hypothesis:
Based on the data we’ve collected and emerging evidence in the field, we believe that cancer treatment can no longer be approached as an individual entity. Next-generation drugs must take into account the various components of the microenvironment and their dynamic changes.
Our Approach:
Our goal is to develop a next-generation therapeutic system that targets the entire tumor microenvironment by analyzing and anticipating the dynamic communication among cells that promote tumor growth and resistance to current treatment methods. To achieve this, we utilize state-of-the-art technologies to reveal the cellular communication occurring between cells and construct sophisticated computational models to depict the complex intercellular interactions.
Our Discoveries:
Location Drives Tumor States: We have developed a novel computational algorithm that utilizes the phenomenon of “spatial-lag” to investigate the relationship between the state of a tumor and its location. The results of this analysis revealed that several tumor cell communities were strongly influenced by their proximity to immune cells. Our findings provide compelling evidence to support the notion that stromal cells, through both cell-cell contact and paracrine signaling, can significantly impact the intrinsic state of tumor cells. These observations align with a growing understanding in cancer research that tumor heterogeneity is not exclusively driven by genetic mutations, but also by epigenetic alterations that arise from the interactions between cancer cells and normal cells in the surrounding environment. Our research has important implications for understanding the varied response to targeted drugs, as our analysis has demonstrated that the degree and type of immune cell infiltration into a tumor can greatly contribute to the complexity of tumor cell heterogeneity, which poses a significant challenge for targeted therapies.
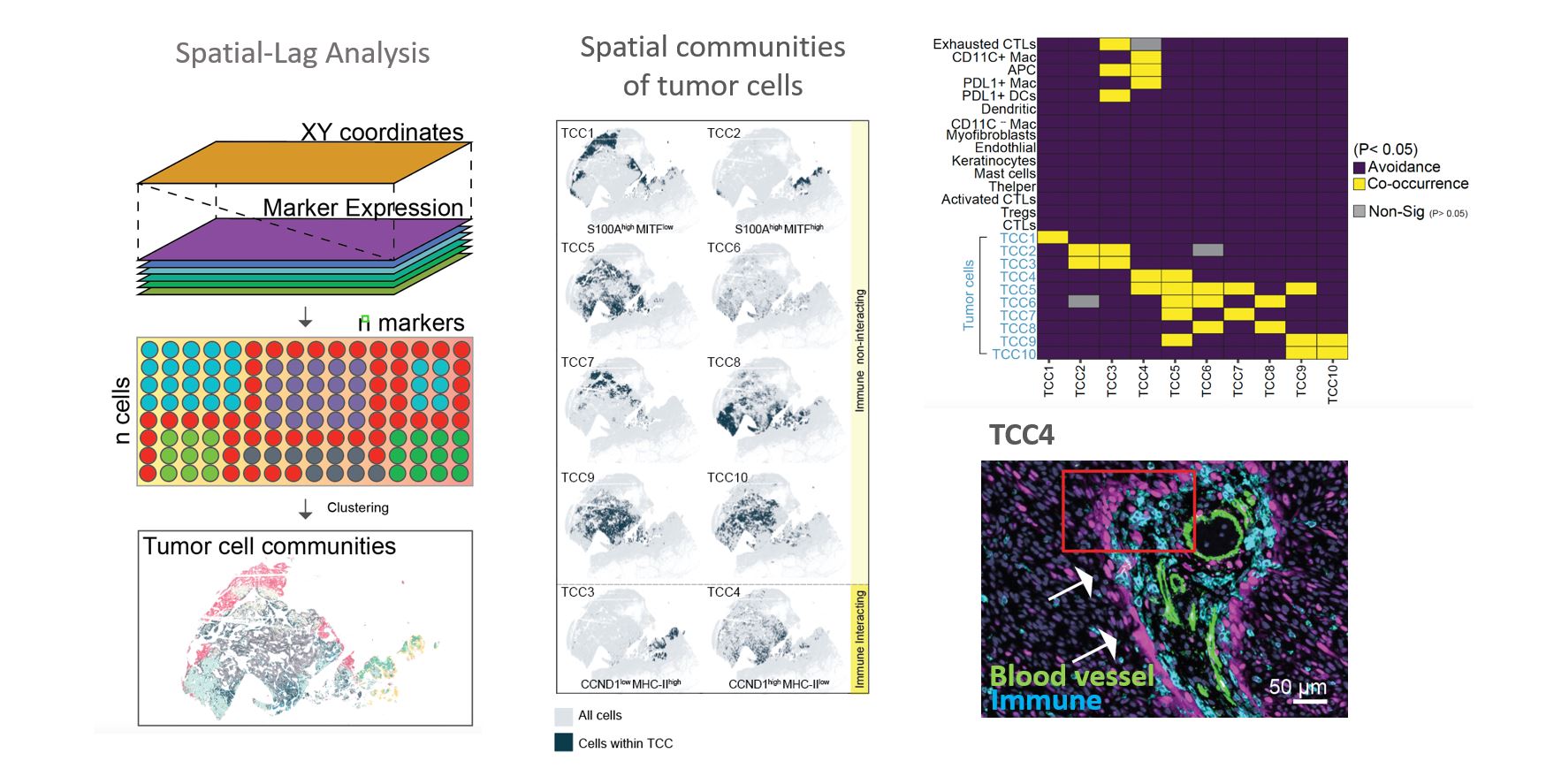
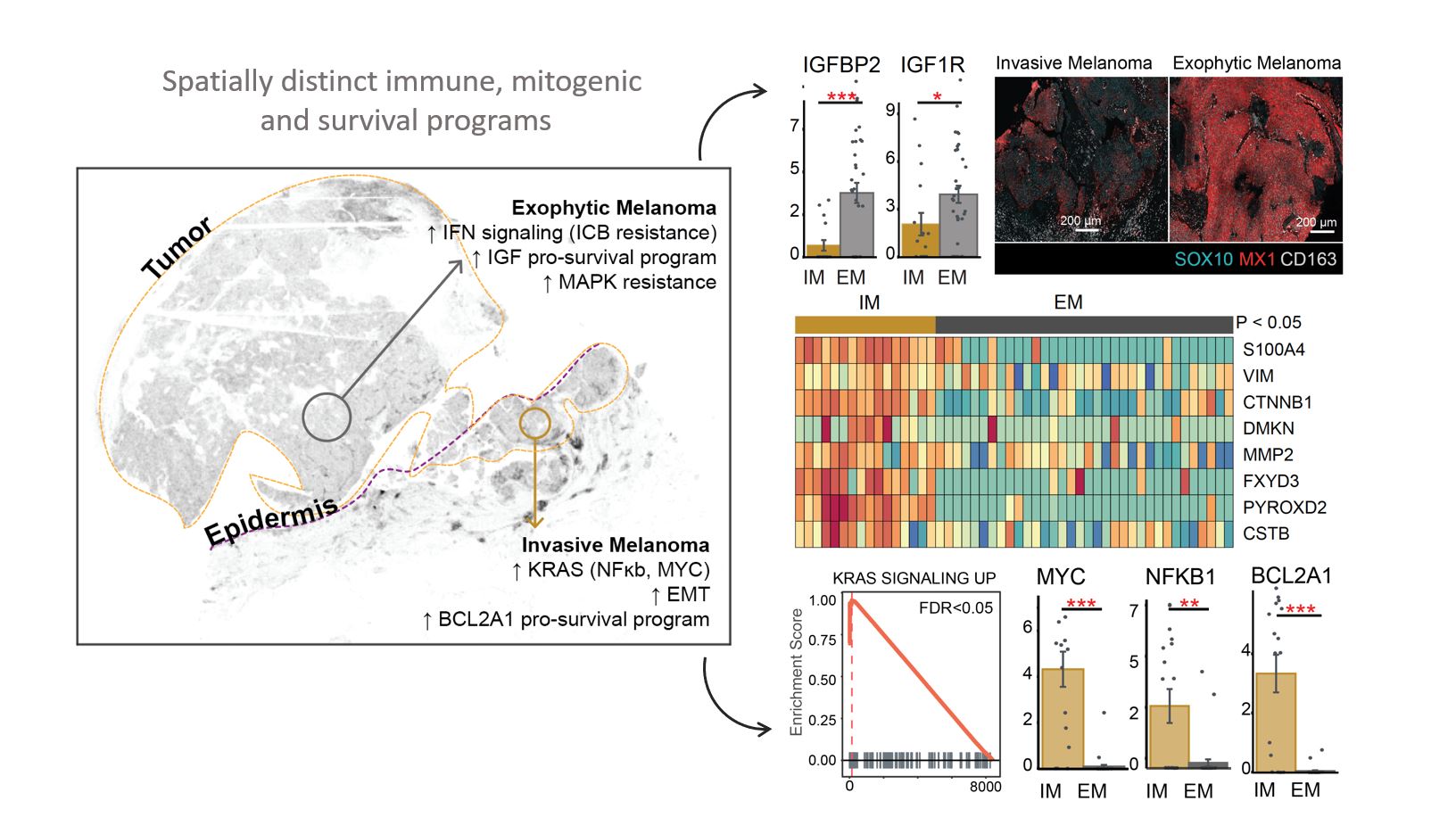
Spatially Distinct Pathways between Tumor Regions: We investigate whether differences in tumor state are driven by alterations in a few markers or if entire pathways are being rewired. To address this question, we employed micro-region sequencing. Our findings indicate the activation of vastly different pathways in tumor cells located only a few millimeters apart within a single melanoma specimen. Specifically, the invasive tumor region exhibited upregulation of KRAS signaling, MYC, NFKB, genes involved in epithelial to mesenchymal transition, and anti-apoptotic survival mechanisms. In contrast, tumor cells in the exophytic region did not show activation of these programs but rather exhibited upregulation of a different set of programs, including insulin-like growth factor signaling and differential expression of interferon signaling. These results are of great significance, as they suggest that a particular drug may be effective in one region but not in another. For example, we identified markers of MAPK resistance in one region but not in the other. Additionally, we found that MX1, which has been linked to resistance to immunotherapy through a PDL1-independent epigenetic mechanism, was upregulated in one region compared to the other.
Recent advancements in imaging technologies have enabled the capture of large-scale and high-dimensional data of biological samples. With these technological advancements, we have access to detailed information about the spatial organization of cells and their interactions within the tissue microenvironment. However, the analysis of such data poses a significant challenge, particularly when it comes to making sense of the vast amounts of information present in multiplexed imaging and spatial transcriptomics data.
To overcome these challenges, we are turning to artificial intelligence and machine learning methods. These techniques offer powerful and efficient tools to process and analyze large-scale and high-dimensional data. By leveraging AI/ML methods, we can gain insights into the encoded cellular neighborhood information of biological samples and the molecular interactions that occur within the tissue microenvironment. In this context, this article explores the potential of AI/ML methods for analyzing complex imaging data, particularly using multiplexed imaging data and spatial transcriptomics data, and their implications for advancing our understanding of biological systems.
Our Algorithms and Tools:
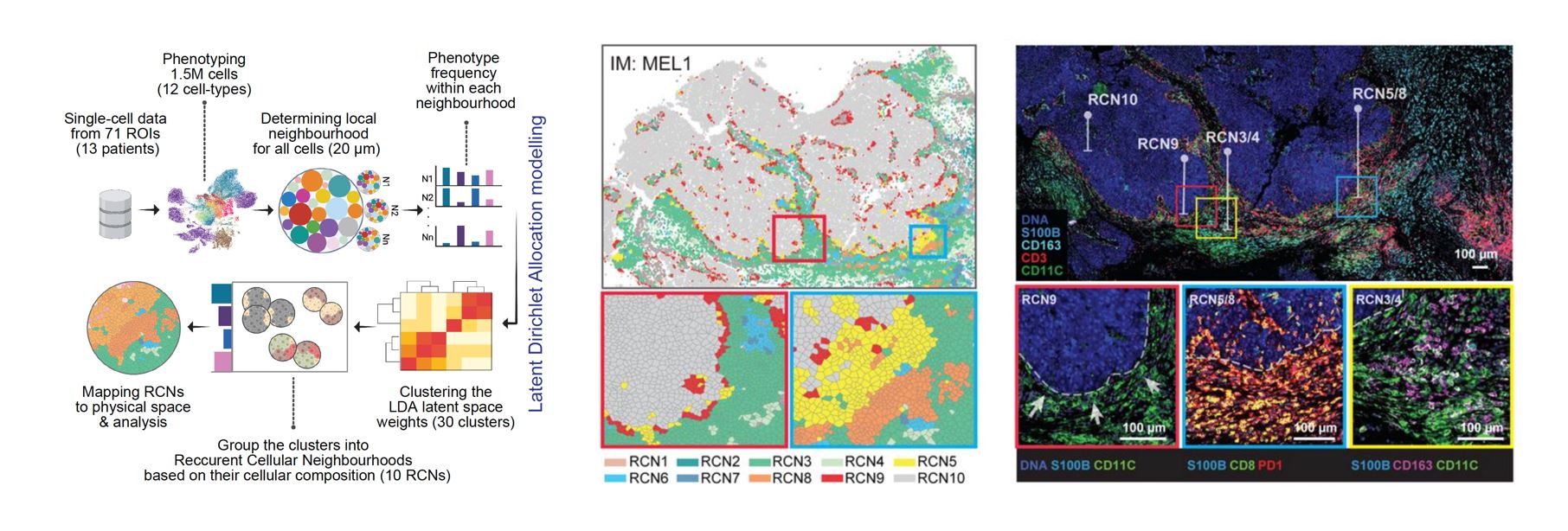
Recurrent Cellular Neighborhoods: To capture and quantify the patterns present in the complex imaging data, we developed a computational algorithm that utilizes latent Dirichlet allocation (LDA) modeling. This method, commonly used in computer vision and natural language processing, allows for the grouping of similar objects based on co-occurrence of certain features. To illustrate this, let’s consider a hypothetical scenario in which we have 10,000 different books with no prior knowledge of their contents and we want to categorize them into meaningful topics. LDA would count the usage of words in each of the books and group them into abstract topics based on co-occurrence of similar words. In our case, instead of books, we have cell neighborhoods, and instead of words, we have cell types. The LDA method counts the occurrence of each cell type in each neighborhood and groups them into abstract topics based on the co-occurrence of similar cell types.
The complexity of the cellular neighborhood can be appreciated with two examples applied to a melanoma tumor sample. The first example is RCN9, which captures the tumor-stromal interface, highlighted in red, enabling us to explore the types of cells present in this region. Through the analysis, we found that the majority of cells present in the tumor-stromal interface were activated myeloid cells. The second example is RCN5, highlighted in yellow, which we identified as an immunosuppressive niche with functional significance. The LDA method allows us to identify such regions of interest and investigate the cellular composition and interactions within them.
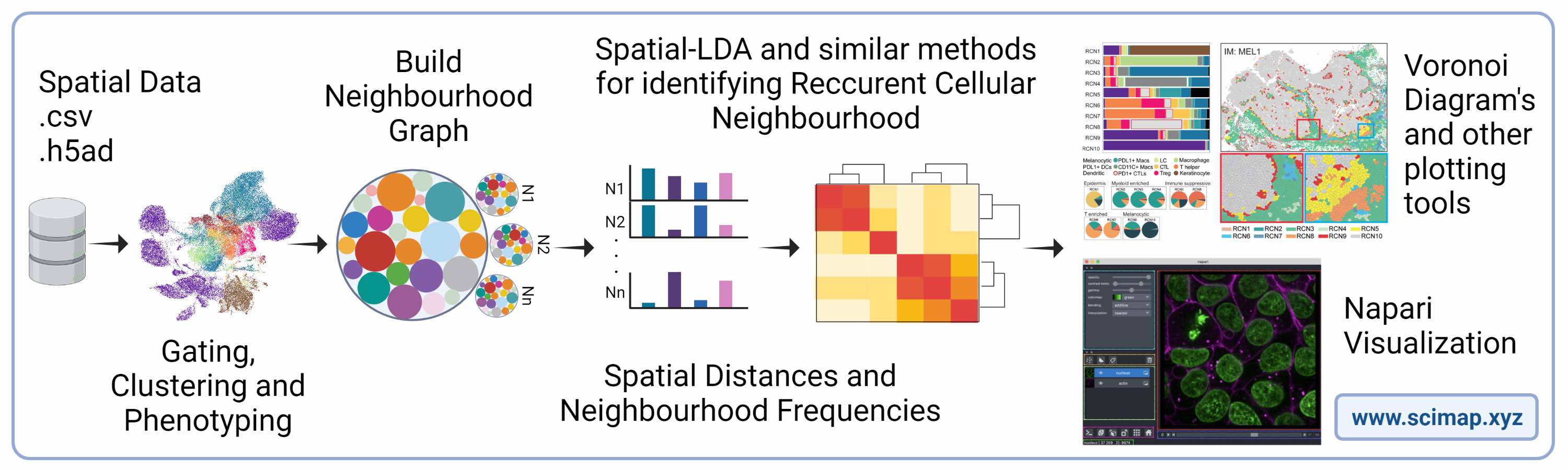
SCIMAP: Spatial Single-Cell Analysis Toolkit: Scimap is a software toolkit that can effectively analyze spatial molecular data in a scalable manner. Its underlying framework is capable of processing various types of spatial datasets mapped to XY coordinates. The toolkit is built on the anndata framework, which facilitates easy integration with other widely used single-cell analysis toolkits. Scimap offers a broad range of functionalities including preprocessing, phenotyping, visualization, clustering, spatial analysis, and differential spatial testing. Its Python-based implementation is highly efficient, enabling the analysis of large datasets that contain millions of cells.
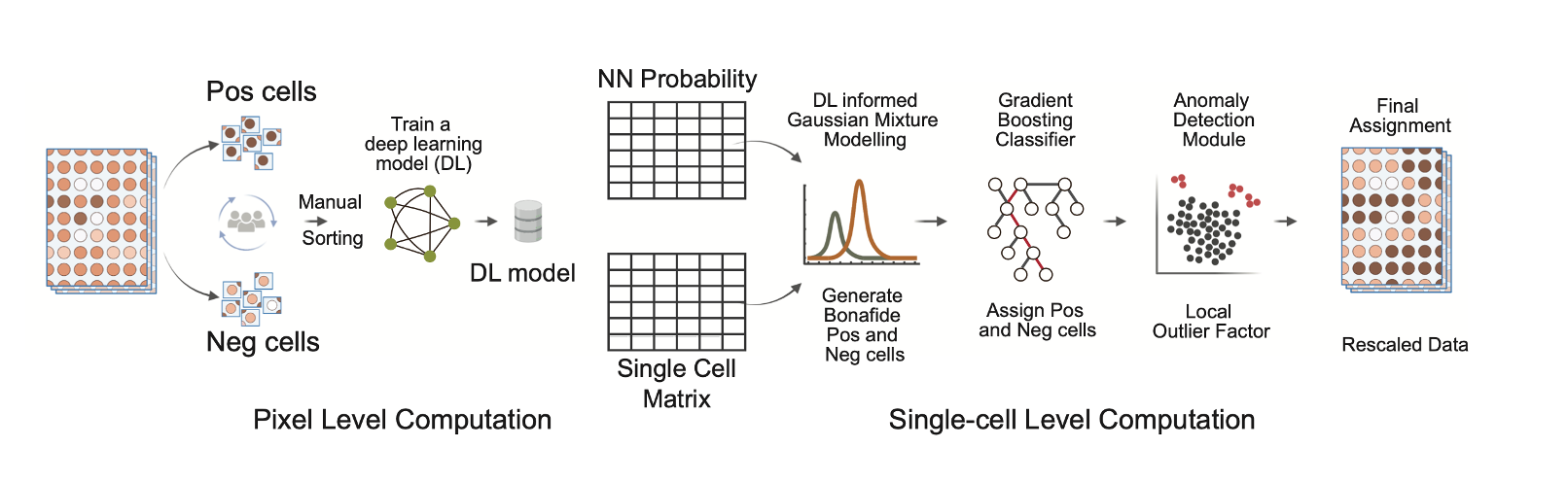
GATOR: A scalable framework for automated processing of highly multiplexed tissue images: Multiplexed imaging has greatly improved our understanding of spatial biology and cell communication networks in tissue development and diseases. But the challenge of scalable application remains due to the need for human involvement. We created GATOR, a computational framework that combines deep learning and statistical approaches to pre-process and phenotype single cells without human intervention. GATOR accurately learns intercellular protein expression patterns and can handle artifactual images. An end-to-end Python implementation of GATOR is available, making it easier to apply spatial biology in large-scale clinical and research settings.
Recent advances in multiplexed imaging have enabled the visualization of the tumor microenvironment with subcellular resolution, providing new opportunities to study the complex interactions between different cell types in 3D. In this context, we are developing new techniques to reconstruct and analyze the 3D structure of the tumor microenvironment in melanoma patient samples. Our approach allow us to study the spatial organization and interactions of various cells in the tumor. By leveraging the power of multiplexed imaging, we hope to gain a more comprehensive understanding of the tumor microenvironment at sub cellular resolution.
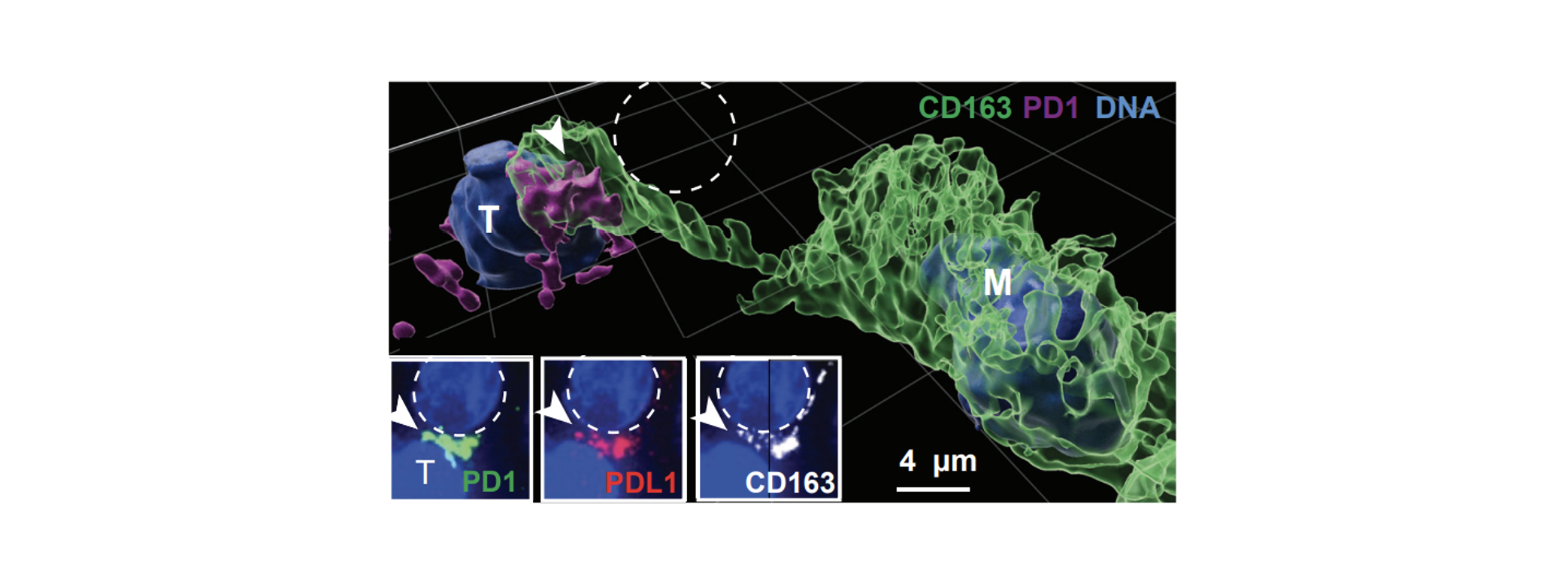
PD1-PDL1 interaction: In the early stages of tumor development, we observed an immune-suppressive interaction between PDL1 ligands on macrophages and dendritic cells, and PD1 positive T cells. We found that PDL1 positivity on myeloid cells was spatially limited to those in close proximity to the tumor, creating a wall-like structure between killer T cells and tumor cells. This arrangement is believed to establish a highly localized immune suppressive microenvironment that shields vulnerable tumor cells from cytotoxic T cell attacks. The image illustrates the ligand-receptor interaction between PDL1+ macrophages and PD1+ T cells at the boundary between tumor cells and stroma.